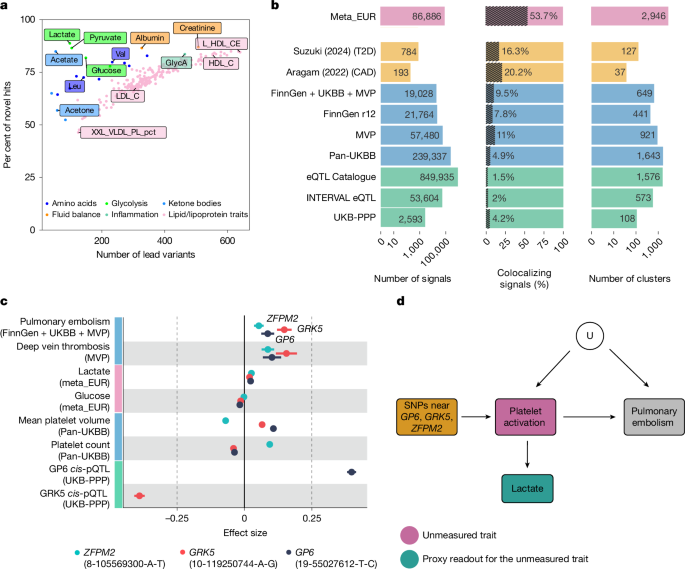
Large-scale GWAS of metabolic traits have identified new associations, but for more than half of NMR-captured metabolic traits, genome-wide significant variants explain less than half of heritability, requiring larger sample sizes. Existing studies using the Nightingale Health NMR platform have mostly focused on common variants and exome sequencing, leaving low-frequency genome-wide variation largely unexplored. Larger studies also increase challenges in interpreting associations, especially when variants show pleiotropy across correlated metabolic traits, raising concerns about naive use in Mendelian randomization. GWAS were performed for 249 metabolic traits in the Estonian Biobank and multiple UK Biobank ancestry groups. Population-specific imputation panels enabled testing 10–96 million variants, stratified by minor allele frequency into common, low-frequency, and rare categories.
"Recent large-scale GWAS of metabolic traits have continued to uncover novel associations and biological insights8,9,10,11,12,13,14. However, for more than half of the metabolic traits that are captured by nuclear magnetic resonance (NMR) spectroscopy, the proportion of heritability explained by genome-wide significant variants remains below 50% (ref. 12), indicating that much larger sample sizes are needed to identify the remaining genetic effects."
"Furthermore, most existing association studies using the Nightingale Health NMR platform have been limited to common variants8,9,10,12 and exome sequencing13,15, leaving the full genome-wide spectrum of low-frequency genetic variation unexplored. Finally, larger sample sizes and increased statistical power also bring new challenges for interpreting genetic associations, particularly when genetic variants have pleiotropic effects on several correlated metabolic traits."
"In particular, there is a growing concern that naive use of these associations in the Mendelian randomization16 framework can lead to spurious and misleading findings17,18. Association testing and meta-analysis We performed GWAS for 249 metabolic traits (Supplementary Table 1) in the Estonian Biobank (EstBB; n = 185,352) and 6 genetic ancestry groups from the UK Biobank (UKBB; n = 434,020) (Extended Data Fig. 1)."
"Relying on the population-specific genotype imputation panel for the EstBB20 and the Genomics England21 and TopMed22 imputation panels for the UKBB allowed us to test 10-96 million variants across genetic ancestry groups (up to nine times more than previous studies using the same NMR platform). On the basis of minor allele frequency (MAF), we stratified these variants into three bins: common variants (MAF > 1%), low-frequency variants (MAF between 0.1% and 1%) and rare variants (MAF < 0.1%)."
Read at Nature
Unable to calculate read time
Collection
[
|
...
]